.jpg)
药物生产过程微生物限度检查方法的改进与准确性验证研究

王志杰
遂成药业股份有限公司 河南新郑 451100
引言
药物的质量直接关系到患者的健康和生命安全,而微生物污染是影响药物质量的重要因素之一。在药物生产过程中,对微生物限度进行严格检查是确保药物安全有效的关键环节。传统的微生物限度检查方法虽然在一定程度上能够检测出药物中的微生物污染情况,但随着药物生产技术的不断发展和对药物质量要求的日益提高,其局限性逐渐显现。因此,对药物生产过程微生物限度检查方法进行改进并验证其准确性具有重要的现实意义。
1 传统微生物限度检查方法的局限性
1.1 操作繁琐
传统的微生物限度检查方法通常需要经过多个步骤,包括样品的稀释、接种、培养以及计数等。每个步骤都需要严格按照规定的操作流程进行,稍有不慎就可能导致实验结果不准确。例如,在样品稀释过程中,如果稀释倍数不准确,就会影响后续的微生物计数结果;在接种过程中,如果接种量不均匀,也会导致培养后的菌落数不能真实反映样品中的微生物含量。
1.2 检测周期长
传统的检查方法需要较长的培养时间才能观察到微生物的生长情况。一般来说,细菌需要培养1-3 天,真菌需要培养3-5 天甚至更长时间。较长的检测周期使得药物生产企业无法及时获得微生物污染的信息,难以及时采取措施控制污染,从而增加了药物质量风险。
1.3 准确性受多种因素干扰
传统方法的准确性容易受到多种因素的影响,如培养基的质量、培养条件(温度、湿度、氧气含量等)、操作人员的技术水平等。例如,培养基的质量不佳可能导致微生物生长不良或出现假阳性、假阴性结果;培养条件不稳定会影响微生物的生长速度和形态,给计数带来困难;操作人员的技术水平参差不齐,也会在一定程度上影响实验结果的准确性。
2 微生物限度检查方法的改进思路
2.1 引入新型检测技术
微生物检测领域的创新技术正逐步革新传统方法。除常规的流式细胞术和PCR 外,等温扩增技术(如LAMP、RPA)可实现无需热循环的快速核酸检测,尤其适用于现场或资源有限的环境。数字PCR(dPCR)通过微滴分区实现绝对定量,显著降低基质干扰,适用于复杂样品中的低丰度微生物检测。高通量测序(如16SrRNA/ITS 测序)可同时分析样品中的多种微生物,适用于污染溯源和未知菌种筛查。
2.2 优化样品处理流程
样品前处理直接影响微生物回收率和检测准确性。膜过滤法可结合表面修饰技术(如亲水/疏水膜)提升目标微生物的截留效率,而超声辅助提取或酶解法可增强生物膜或包埋微生物的释放。对于含防腐剂或抗生素的样品,可采用中和剂(如硫代硫酸钠、卵磷脂)消除抑菌效应。自动化样品处理系统(如液体工作站)可标准化稀释、混匀及接种步骤,减少人为误差。
2.3 建立标准化操作流程
标准化操作流程(SOP)的制定需基于风险评估和方法验证数据,涵盖样品采集、运输、储存、前处理、检测及数据分析的全过程。关键控制点(如无菌操作、培养条件、阳性对照设置)需明确规定,并建立偏差处理机制。SOP 应定期审核并更新,以适应技术迭代或法规变化。配套的质量控制(QC)措施应包括环境监测(如沉降菌测试)、培养基适用性检查及人员能力验证。
3 改进后方法的准确性验证
3.1 验证指标的选取
准确性验证需要选取合适的指标来评估改进后方法的可靠性。常用的验证指标包括灵敏度、特异性、准确度、精密度等。灵敏度是指方法能够检测出的最低微生物含量,反映了方法对微量微生物的检测能力;特异性是指方法能够准确区分目标微生物和非目标微生物的能力;准确度是指测量结果与真实值之间的接近程度;精密度是指多次测量结果之间的一致性。
3.2 验证实验的设计
3.2.1 灵敏度验证
灵敏度验证的核心在于建立检测限(LOD)与定量限(LOQ)的可靠标准。实验中需采用梯度稀释法将标准微生物菌株制备成 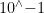 至10∘⋅7CFU/mL 系列浓度,每个浓度设置至少 6 个平行样本。关键控制点包括:菌液活细胞比例的流式细胞仪确认、基质效应的消除(如通过PBS 缓冲液模拟样品本底),以及抑制物的干扰测试。验证过程中需记录假阴性率随浓度降低的变化趋势,当假阴性率 55% 时对应的浓度即为方法检测限。为提高结果可信度,建议采用国际标准菌株(如ATCC 系列),并同步进行核酸提取效率验证(如添加内标基因)。
至10∘⋅7CFU/mL 系列浓度,每个浓度设置至少 6 个平行样本。关键控制点包括:菌液活细胞比例的流式细胞仪确认、基质效应的消除(如通过PBS 缓冲液模拟样品本底),以及抑制物的干扰测试。验证过程中需记录假阴性率随浓度降低的变化趋势,当假阴性率 55% 时对应的浓度即为方法检测限。为提高结果可信度,建议采用国际标准菌株(如ATCC 系列),并同步进行核酸提取效率验证(如添加内标基因)。
3.2.2 特异性验证
特异性验证需构建系统化的交叉反应评价体系。实验设计应涵盖以下维度:1)近缘菌种(如大肠杆菌VS 志贺氏菌);2)相同科属的干扰菌(如金黄色葡萄球菌 VS 表皮葡萄球菌);3)样品中常见共生微生物。检测层面需结合形态学观察(如革兰氏染色)、生化反应(API 试条)和分子标记(特异性引物熔解曲线分析)。对于免疫学方法,还需验证类风湿因子、异嗜性抗体等干扰物质的影响。数据分析可采用受试者工作特征曲线(ROC)计算 AUC 值, >0.9 表明方法具有显著区分能力。
3.2.3 准确度和精密度验证
准确度验证应采用标准参考物质(SRM)或能力验证(PT)样本作为基准,通过配对 t 检验分析测量均值与真值的偏差。对于微生物检测,需特别关注Log 标度下的误差分析,建议采用Bland-Altman 法评估一致性界限。精密度验证应覆盖三种水平:1)重复性(同操作者/设备/日内);2)中间精密度(不同日期/实验员);3)重现性(跨实验室)。实验设计需遵循嵌套方差分析原则,将总变异分解为操作者间、批次间、运行间等分量。
结束语
本文对药物生产过程微生物限度检查方法进行了改进,并对其准确性进行了验证。通过引入新型检测技术和优化样品处理流程,改进后的方法在操作便捷性、检测效率和准确性方面都有了显著提高。准确性验证实验结果表明,改进后的方法具有较高的灵敏度、特异性和准确度,精密度也符合要求,能够更好地满足药物生产质量控制的需求。药物生产企业可以采用改进后的方法进行微生物限度检查,以提高药物质量,保障患者的用药安全。
参考文献
[1]李待军,罗飞,王引弟,等.补骨脂酊微生物限度检查方法适用性试验研究[J].甘肃中医药大学学报,2025,42(04):52-56.
[2]曾娜霞,吴狄.药食同源龙眼肉微生物限度检查方法适用性研究[J].轻工科技,2025,41(04):16-19+34.
[3]杨晓峰,秦利芬,李聃,等.2 种外用制剂微生物限度检查方法的建立[J].现代医药卫生,2025,41(07):1555-1559.
[4]郑敏,武鑫,王小宁,等.两种中药骨科外用散剂的微生物限度方法适用 性考察[J].实验室检测,2025,3(11):193-195.
[5]张利仂,肖文斌,钟志坚,等.含抑菌成分中药片剂的微生物限度检查方法学研究[J].工业微生物,2025,55(02):22-24.
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)