.jpg)
自动电位滴定法 氧化物中主成分测定的实验研究

宁志坚 王小维 张瑞娟 李露 刘云河
四川红华实业有限公司 四川峨眉山 614200
1、引言
日常分析工作中我厂需要对氧化物中主成分进行测定,随着分析任务的增加,原分析方法,分析时间长,人为因素影响大,易出现滴定突跃不明显,终点难于辨认,分析结果重现性差等情况日趋显著。因此,为及时有效的完成分析任务,本文研究开展了自动电位滴定法测定金属氧化物中主成分含量的分析方法实验研究工作。通过滴定模式选择,滴定参数设定,方法精密度、准确性验证等实验工作,最终确定了仪器的最优工作参数,完善了滴定分析条件,为今后分析提供了可靠的质量保证。当置信度为 95% 时,取样量为 1.0 克的金属氧化物,所建方法的相对标准偏差优于 ±0.025% 。
2、实验部分
2.1、方法原理
氟化物经水解、蒸干、灼烧转化成氧化物固体,将固体氧化物用磷酸溶解,在磷酸介质中用硫酸亚铁还原,在合适的温度下以钼(Ⅵ)作催化剂,用硝酸氧化过量的亚铁,加氨基磺酸除去氧化过程中产生的氮氧化物。在硫酸氧钒的存在下,用重铬酸钾溶液滴定至终点,由此精确计算出氧化物中的主成分。
2.2、试剂
除非另有说明,本文所使用的试剂,均为符合国家标准的分析纯试剂,分析用水均为去离子水。
2.2.1、硫酸( H2SO4 )。
2.2.2、硫酸 (H2SO4):( 1+1)
2.2.3、硝酸( HNO3 )。
2.2.4、磷酸( H3P03 )。
2.2.5、硫酸亚铁( FeSO4 )溶液: ρ=280g/L
称取硫酸亚铁 28g±1g 于 50mL 烧杯中,加 50mL 水、 10mL 硫酸(2.2.1),不断搅拌至全部溶解,转移至 100mL 容量瓶中,用水定容,摇匀。此溶液不稳定,有效期为1 周。
2.2.6、氨基磺酸 (NH2SO3H) 饱和溶液。
2.2.7、氧化剂:硝酸 - 氨基磺酸- 钼酸铵混合溶液。
称取 1.0g 钼酸铵【( NH4 ) Γ6M0.O24.4H2O 】溶于 100mL 水中,加125mLHNO3 (2.2.3),冷却,再加入 25mL 氨基磺酸饱和溶液(2.2.6),摇匀,保存在试剂瓶中。此溶液不稳定,有效期为1 周。
2.2.8、硫酸氧钒( VOSO4⋅H2O )溶液: 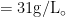
称取 3.1g 无水硫酸氧钒,用 5mLH2SO4 (2.2.1)溶解,并稀释至100mL ,保存在试剂瓶中。此溶液不稳定,有效期为1 周。
2.2.9、重铬酸钾( K2Cr2O7 )固体:标准物质GBW06105。
使用前在( 130%±10 ) C 烘 6h,取出后置于干燥器中,冷却至室温,备用。
2.2.10、重铬酸钾( K2Cr2O7 )溶液: 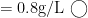 。
。
称取 0.8g 重铬酸钾(2.2.9),精确到 0.1mg ,用水溶解,转入1000mL 容量瓶中,用水定容,摇匀。
2.3、仪器
2.3.1、自动电位滴定仪,梅特勒 T50 型。
2.3.2、分析天平,分度值为 0.1mg
2.3.3、铂环复合电极,DM140-SC 型。
2.3.4、电磁搅拌器,带有氟塑料包裹的搅拌子。
2.4、实验步骤
2.4.1、将氟化物经水解、蒸干、灼烧转化成氧化物固体作为试样。
2.4.2、用分析天平称取约 1g 固体试样(2.4.1),精确至 0.1mg ,记录质量为 m1 ,置于 400mL 高型烧杯中,沿壁加入磷酸(2.2.4)30mL,滴加(2~3)滴重铬酸钾溶液(2.2.10),在不断摇动下,加热至试样完全溶解(控制温度使溶液不得沸腾),冷却至室温。
2.4.3、计算试样中的理论主成分含量,用 ω 表示。按式(1)计算称取重铬酸钾固体(2.2.9)理论需要量 m 。实际称取质量比理论需要量少( 4~8g ),精确至 0.1mg ,记录质量为 m2 ,备用。
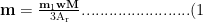
式中:
m—重铬酸钾固体(2.2.9)理论需要质量,g;
m1 —试样的质量,g;
w—试样中主成分的理论含量的质量分数;
M—重铬酸钾的相对分子质量;
Ar—平均相对原子质量。
2.4.4、将烧杯置于电磁搅拌器上,在搅拌情况下,沿壁加入 5mL氨基磺酸饱和溶液(2.2.6)。
2.4.5、加入 8mL 硫酸亚铁溶液(2.2.5)。硫酸亚铁溶液应通过移液管直接加入到试样溶液中,不得沿烧杯壁流下。在室温下继续搅拌还原不得少于 1min 。(以下步骤应连续进行)。
2.4.6、沿壁加入 5mL 硫酸(2.2.2),调节溶液温度在(35~40) C ,再沿烧杯壁加入 8mL 氧化剂(2.2.7),搅拌( 2~3)min 后,再放置 0.5min 。(加入氧化剂后,试样溶液变成棕黑色,此颜色应在30s 左右消失)。
2.4.7、在搅拌条件下,沿壁加入水 90mL 水, 10mL 硫酸氧钒溶液(2.2.8)。(为了避免显著性偏差,2.4.7~2.4.9 步骤应在加入硫酸氧钒溶液后 7min 内完成。)
2.4.8、将事先称取好的固体重铬酸钾(2.4.3)完全转入试样溶液(2.4.7)中。
2.4.9、设置自动电位滴定仪(已预热)程序,开始用重铬酸钾溶液滴定(2.2.10)。记录消耗重铬酸钾体积(V)。
2.5、计算结果
氧化物中主成分含量按式(2)计算:
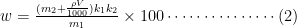
式中:
w—氧化物中主成分的质量分数 数值以百分数表示;
m2 —重铬酸钾固体(2.2.9)的质量,g;
ρρσ(ρρσ)=ρρ(ρρσ)ρρ(ρρσ) —重铬酸钾溶液(2.2.10)的质量浓度, g/L ;
V—等当点时所消耗重铬酸钾溶液(2.2.10)的体积,mL;
k1 —转换因子, k1=2.4274 ;
k2 —浓缩因子, 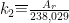 ;
;
m1 —称取的氧化物试样的质量, g
3、结果与讨论
3.1、滴定剂添加模式确定
T50 型自动滴定仪提供的滴定模式有动态法(DYN), 增量法(INC)两种模式,动态法可靠性强,反应速度快,滴定突跃明显,为此本文选择动态添加(DYN)模式。
3.2、预搅拌时间与速度的确定
加入重铬酸钾固体,为使其充分反应,需预搅拌一定时间,观察电位变化,当电位值稳定,说明重铬酸钾固体充分反应。实验表明,设定预搅拌时间为 10S,满足实验要求。搅拌速度对测量结果准确性影响较大,若速度过低,反应不完全,从而使电极平衡速度慢,终点退后;若速度过高,电极表面产生气泡,不能得到稳定信号,对结果影响极大。因此适宜的搅拌速度应根据溶液性质及加入量设定,在搅拌过程中产生尽可能大的漩涡而又不产生气泡。试验表明,搅拌速度设定为 70% 时,满足实验要求。
3.3、阈值的确定
阈值的大小取决于测试样品的种类、滴定剂种类、滴定剂浓度等因素,一般很难设定正确的阈值,其大小一般为峰高度的 50%~70% 。在完成首次滴定的情况下,在滴定电位范围内采用一阶微商法 ΔE/
ΔV 对滴定剂的体积(V)作图,通过实验,最终设定阈值为 600mv/ mL 。
图 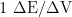 对滴定剂的体积曲线图
对滴定剂的体积曲线图
3.4、终点的确定
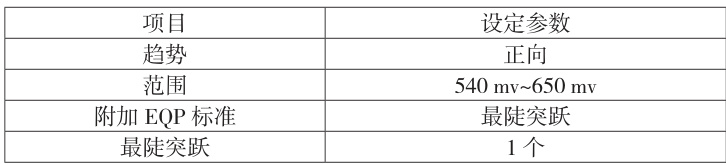
本文研究的滴定类型为等当点滴定,干扰因素较多,可不再设定EQP,选择在一定的范围内电位变化中产生最陡的峰为终点。设定结果见表1。
表1 T50 自动电位滴定仪滴定参数设定表
3.5、精密度试验
称取约 1g 固体试样于 400mL 高型烧杯中,按 2.4.1~2.4.9 步骤进行操作。实验结果见表2。
表2 自动电位滴定法精密度试验数据统计表
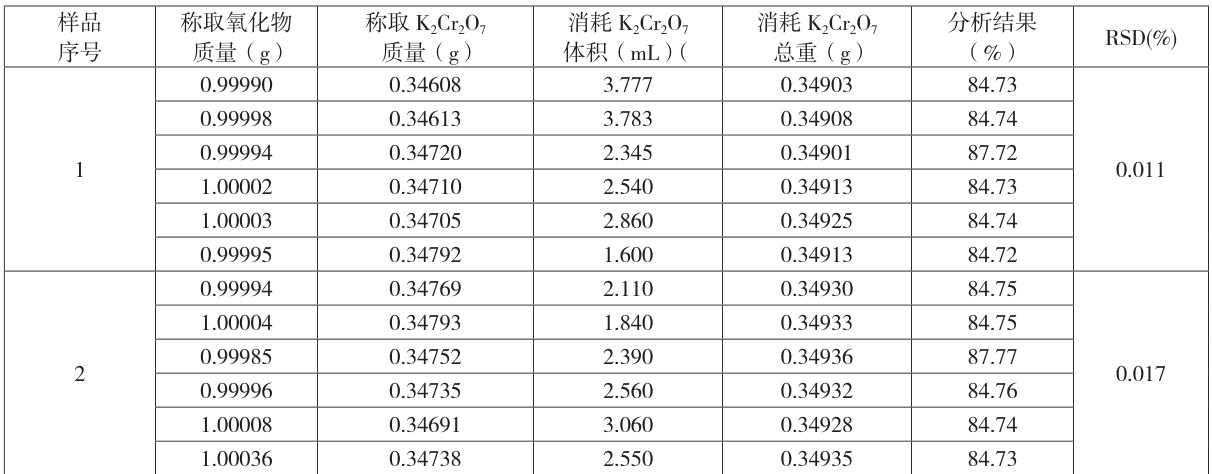
表2 结果表明:自动电位滴定仪参数设置合理,测试结果精度好,分析结果的相对标准偏差优于 ±0.025% ,满足要求。
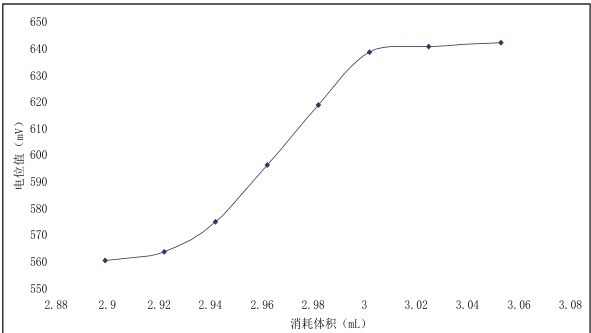
重铬酸钾对 1g 试样进行滴定,实验条件经过优化,滴定剂浓度为 0.8g/L 时,滴定曲线见图 2。同一浓度的滴定剂滴定时,滴定的电位突跃和斜率基本相同。
图2 自动电位滴定曲线图
3.6、准确度试验
称取约 1g 基准氧化物于 400mL 高型烧杯中,按 2.4.1~2.4.9 步骤进行操作。实验结果见表3。
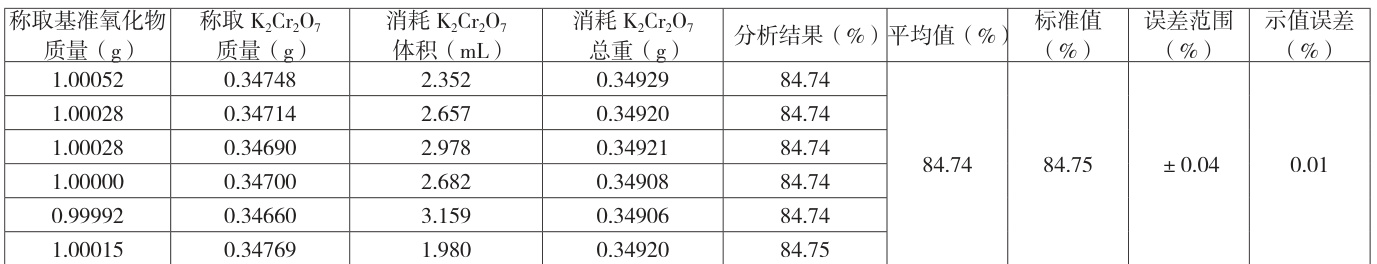
表 3 结果表明:此方法分析结果准确度高,满足我厂分析技术要求,因此电位滴定仪分析方法是准确、可靠的。
称取约 1g 固体试样于 400mL 高型烧杯中,按 2.4.1~2.4.9 步骤进行操作,实验结果见表4。
3.7、方法的应用
表3 自动电位滴定法准确度试验数据统计表
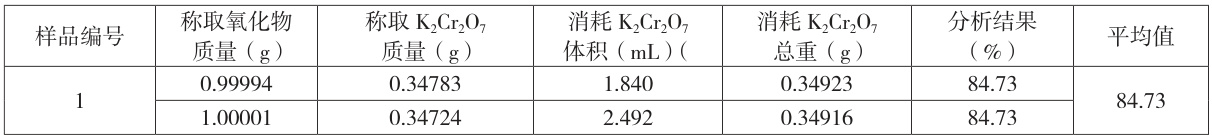
表4 自动电位滴定法应用于实际生产中分析数据统计表
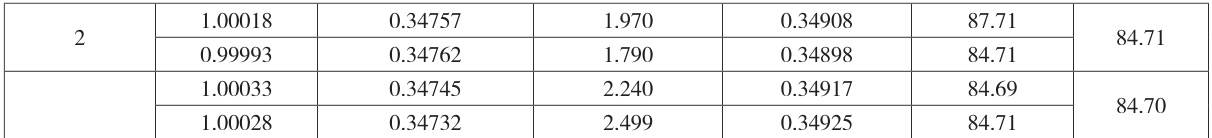
表 4 结果表明:自动电位滴定法在生产中完全能满足氧化物中主成分含量的测定。
3.8、手动滴定与自动电位滴定法滴定比对试验
称取约 1g 基准氧化物 400mL 高型烧杯中,按 2.4.1~2.4.9 步骤进行操作,两种滴定方法进行比对试验,实验结果见表5。
表5 两种方法的比对试验数据表
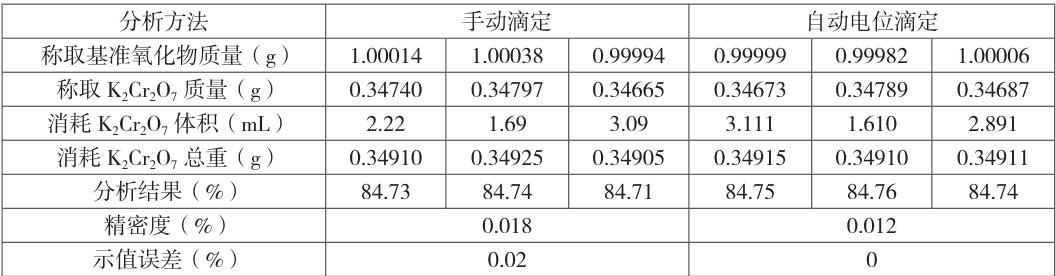
表 5 结果表明:手动滴定与自动电位滴定法滴定两种方法相比较,自动电位滴定法精密度优于手动滴定。
4、结论
利用自动电位滴定仪建立了测定氧化物中主成分测定的方法,该方法的精密度 ±0.025% 。虽然试验结果表明,手动滴定和自动电位法均能满足我厂分析要求,但自动电位法精密度明显好于手动滴定,同时自动电位法避免了手动滴定对人员要求高、终点不易判断、不易实现自动化等缺点。上述两种方法综合比较见表 6
表6 手动滴定与自动滴定比较

参考文献:
[1] 刘 权 卫 . 自 动 电 位 滴 定 仪 测 定 . 原 子 能 科 学 技术 ,2007.9,41(5);546~549. .
[2] 佟琦 , 高丽华 . 莫尔法与自动电位滴定法测定水中氯离子含量的比较. 工业水处理, ,2008,28(11):69~71 .
[3] 董丽丽 . 自动电位滴定法快速测定 钾盐中的钾含量 . 分析试验室 ,2004,23(1):83 ~ 84.
[4] 卢振国 . 自动电位滴定法测定铁矿石中水溶性氯化物 . 理化试验: 化学分册 ,2007,43(9):732~734
[5] 王虹 . 电位滴定法测定铁矿石中水溶性氯化物含量 . 化学分析计量 ,2006,15(1), .43~44 .
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)