.jpg)
肿瘤药物耐药性产生机制及应对策略研究

敖孟霖 杨佳丽通讯作者
云南省肿瘤医院 昆明医科大学第三附属医院 北京大学肿瘤医院云南医院 药学部
引言:肿瘤耐药性分为原发性(治疗前即存在)和获得性(治疗过程中产生)两种,是化疗、靶向治疗及免疫治疗面临的主要挑战,其机制涉及药物外排增强(如 P-gp、BCRP)、药物靶点基因突变(如 EGFR T790M、BCR-ABLT315I)、信号通路代偿激活(如 PI3K/AKT)、表观遗传调控异常、肿瘤干细胞富集及免疫微环境抑制等,深入解析耐药机制并开发有效逆转策略,对改善患者生存预后具有重要意义。本文整合最新研究成果,系统探讨耐药机制及应对策略,为临床转化提供理论支撑[1]。
1. 材料与方法
1.1 研究对象
1.1.1 细胞模型
亲本敏感细胞:A549(非小细胞肺癌)、MCF-7(乳腺癌)、SW480(结直肠癌)耐药株模型:通过梯度浓度递增法建立耐紫杉醇(A549/TAX)、耐顺铂(MCF-7/DDP)、耐西妥昔单抗(SW480/CET)细胞系
1.1.2 动物模型
选用雌性BALB/c 裸鼠(4-6 周龄,体重 18-20g ),将上述耐药细胞( 5×106/ 鼠)皮下注射至小鼠右腋,待肿瘤体积达 100mm3 后开展实验,共设 6 组,每组6 只动物(含对照组)。
1.1.3 临床样本
收集 20 例非小细胞肺癌患者化疗前后的配对肿瘤组织(手术 / 活检样本),所有样本经医院伦理委员会审批,患者签署知情同意书。
1.2 研究方法
1.2.1 耐药性评价
(1)细胞毒性检测:采用CCK-8 试剂盒评估药物敏感性。将细胞接种于96孔板,给予不同浓度药物处理 48 小时,测定 450nm 吸光度并计算 IC50 值。耐药指数(RI)按公式计算:
RI = 耐药细胞 IC50/ 亲本细胞 IC50
(2)细胞凋亡分析:用Annexin V-FITC/PI 双染试剂处理细胞,流式细胞仪检测凋亡率。实验重复3 次[2]。
1.2.2 分子机制研究
(1) 蛋 白 表 达 检 测:qPCR 与 Western Blot 分 析 ABC 转 运 蛋 白(P-gp、MRP1)、凋亡相关蛋白(Bcl-2、Bax)、DNA 修复蛋白(ERCC1)的表达差异。
(2)基因突变筛查:提取细胞及临床样本 DNA,通过二代测序(NGS)靶向检测 EGFR、KRAS 基因突变。
(3)表观遗传调控:采用ChIP-qPCR 技术,分析耐药基因启动子区的组蛋白修饰水平(H3K27ac 激活标记、H3K9me3 抑制标记)。
(4)肿瘤微环境解析:对移植瘤及临床组织切片进行免疫组化染色,定量CD163⁺ M2 巨噬细胞和 FoxP3⁺ Treg 细胞的浸润程度 [3]。
1.2.3 逆转策略验证
(1)体外联合用药:维利帕尼( 5μM )与紫杉醇联用处理A549/TAX 细胞;伏立诺他( 1μM )与顺铂联用处理 MCF-7/DDP 细胞。
(2)体内药效评估:
裸鼠移植瘤实验分四组: 裸鼠移植瘤实验分组:对照组、单药组、联合治疗组,每3 天测量肿瘤长宽,计算体积(公式: V=0.5× 长 × 宽²);记录小鼠生存期 [4]。
1.3 统计分析
数据以均值 ± 标准差表示,采用 GraphPad Prism9.0 软件分析,组间比较用t 检验(两组)或单因素ANOVA(多组), P<0.05 视为具有统计学差异。
2. 结果
2.1 耐药细胞模型的建立与特征
表1 耐药细胞模型特征表

耐药株普遍呈现增殖减缓、凋亡抵抗(如 Bcl-2/Bax 比值升高 2-4 倍)及ERCC1 过表达。
2.2 关键耐药机制解析
(1)药物外排系统激活:A549/TAX 中 P-gp 蛋白表达较亲本提升 5.8 倍( P<0.001 ),钙黄绿素蓄积实验证实外排功能增强。
(2)靶基因突变与通路代偿: 60% 西妥昔单抗耐药样本检出KRAS G12V 突变,并伴随MET 通路磷酸化激活。
(3)表观遗传重塑:MCF-7/DDP 细胞中促凋亡基因 BIM 启动子区 H3K27ac修饰显著降低(ChIP-qPCR, P<0.01 )。
(4)免疫微环境抑制:耐药肿瘤组织内 CD163+ M2 巨噬细胞浸润增加 3 倍
( P<0.001 ),PD-L1 表达同步上调。
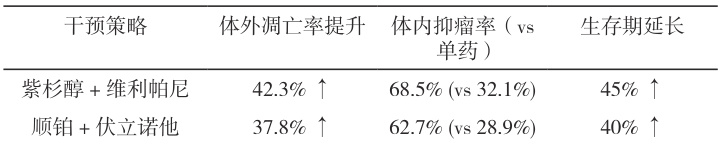
2.3 耐药逆转策略效果
联合治疗显著降低 P-gp 功能( P<0.01 )并逆转 H3K27ac 抑制( P<0.05 ),使得体外凋亡率、体内抑瘤率提升,生存期延长。
表2 联合治疗耐药逆转效果表
3. 讨论
3.1 耐药机制的系统性与动态演化
本研究揭示肿瘤耐药性并非单一静态事件,而是由细胞内在机制、微环境调控与表观遗传重塑共同驱动的动态过程。在细胞层面,发现ABC 转运蛋白(如P-gp )的过表达是化疗药物失效的关键推手。以紫杉醇耐药株 A549/TAX 为例,其 P-gp 表达量激增 5.8 倍,如同在细胞膜上安装了“药物泵”,将化疗药物主动排出细胞外。而在靶向治疗中,基因突变(如 KRAS G12V)和旁路信号激活(MET 磷酸化)则扮演核心角色——当西妥昔单抗阻断 EGFR 通路时,肿瘤细胞通过激活 MET 通路实现“逃生”。微环境的改变同样不容忽视,耐药肿瘤组织中,免疫抑制性细胞(如 CD163+ M2 巨噬细胞和 FoxP3+ Treg 细胞)的浸润显著增加。这些细胞释放抗炎因子(如IL-10、TGF-β),形成一道“免疫屏障”,不仅保护肿瘤细胞免受攻击,还促进 PD-L1 表达,最终导致免疫治疗失效。表观遗传调控的参与为耐药研究提供了新视角,在顺铂耐药的MCF-7/DDP细胞中,促凋亡基因 BIM 的启动子区域组蛋白激活标记(H3K27ac)大幅减少,使该基因处于“关闭”状态。这种沉默是可逆的,解释了为何表观药物能重新唤醒凋亡程序。耐药机制是动态演化的网络系统,治疗压力下,肿瘤细胞通过基因突变、表观适应和微环境重塑实现生存,要求我们采取多维度干预策略。
3.2 联合逆转策略
基于上述机制,探索了两种联合治疗策略,并发现其显著潜力:靶向药物外排泵是经典思路,P-gp 抑制剂维利帕尼与紫杉醇联用时,成功将 A549/TAX细胞的凋亡率提升 42.3% 。其原理是维利帕尼阻断 P-gp 的“泵功能”,使紫杉醇能在细胞内累积并发挥作用。但该策略存在局限——维利帕尼可能引发低血压和心律失常,提示临床需严格监测心血管毒性。表观遗传干预展现出独特优势,HDAC 抑制剂伏立诺他通过增加染色体开放性,重新激活了 MCF-7/DDP细胞中被沉默的凋亡基因(如BIM)。当与顺铂联用时,二者产生协同效应:伏立诺他“解锁”基因,顺铂则诱导 DNA 损伤,最终使抑瘤率提升至 62.7% 。未来突破方向需聚焦三点:(1)动态监测耐药演化:利用 ctDNA 液体活检技术,实时追踪治疗过程中耐药克隆的基因变化,为调整方案提供依据;(2)创新技术干预:CRISPR 筛选可快速鉴定耐药关键靶点;双特异性抗体则能同时阻断主通路和代偿通路,预防“旁路激活”;(3)微生物组调控:最新研究发现,肠道菌群代谢物(如丁酸盐)可影响化疗药物(如环磷酰胺)的疗效,这为联合益生菌干预提供新思路。
综上所述,克服耐药性需整合多组学分析揭示机制网络,并通过“靶向 + 表观”等合理化联合治疗实现协同增效。之后应深入探索微环境与表观遗传的交互作用,推动实验室成果向临床转化。
参考文献:
[1] 徐瑞雪 , 王宇 . 紫草素联合化疗药物逆转肿瘤耐药性的研究进展 [J]. 现代药物与临床 , 2024, 39 (08): 2154-2161.
[2] 王立康, 韩澳迎, 张丽娜. 奥沙利铂在胃癌耐药性中的研究进展[J]. 广东化工 , 2024, 51 (15): 93-94+89.
[3] 徐娇 , 刘雯雯 , 周志军 , 邓豫豫 , 李世洋 , 徐易 , 李昌夏 , 陶小华 . 138 例皮肤肿瘤患者皮损部位细菌培养及耐药性分析 [J]. 药品评价 , 2024, 21 (07): 893-897.
[4] 屈元晔, 肖伟强, 常彦敏, 孙明月, 王小昆, 许青霞. 河南某肿瘤医院临床分离菌分布及耐药性分析 [J]. 中国合理用药探索 , 2024, 21 (07): 42-51.
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)